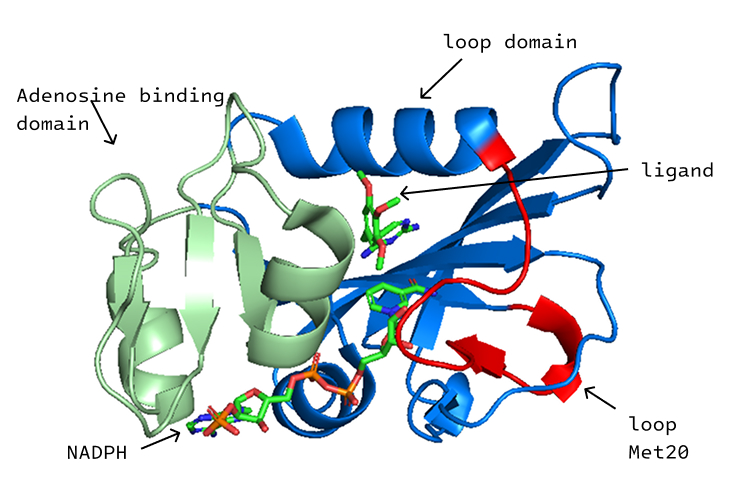
EcDHFR-NADPH-ligand systems
Representative trimethoprim analogues are modelled in the cofactor-bound EcDHFR pocket for comparable trajectory analysis.
A PROTO-NOOS biocomplex-analysis protocol for moving beyond geometric docking scores by integrating molecular dynamics, enhanced sampling, binding energetics, and local electronic-structure descriptors for EcDHFR-NADPH-ligand systems.
Escherichia coli dihydrofolate reductase (EcDHFR) is a validated antibacterial target whose ligand recognition cannot be adequately described by docking scores alone, a limitation shared by any protein-ligand system of comparable complexity.
We present a multi-scale computational protocol designed to characterize EcDHFR-ligand complexes with physicochemical and electronic detail, while generating structured data suitable for downstream machine-learning parameterization. Classical molecular dynamics (MD) simulations assess binding-mode stability, residue-level flexibility, and dominant conformational states of the binding pocket, including its response to structurally diverse ligands. PLUMED-driven metadynamics further probes pocket conformational transitions. MM-GBSA provides comparative binding energetics across selected systems. For representative trajectory frames, CP2K is used to examine the local electronic structure of the binding region, including atomic charge distribution, charge transfer, and polarization effects, phenomena systematically neglected by classical force fields.
Together, these analyses build a domain-informed dataset that links computed descriptors to experimentally known values, enabling iterative refinement of earlier-stage approximations in the pipeline. The methodology is developed for EcDHFR but is designed to generalize to homologous targets.
We argue that integrating structural, energetic, and electronic descriptors is a prerequisite for meaningful in silico candidate selection beyond geometric scoring.
This page records the current poster-stage protocol. The manuscript is not attached yet, so the abstract is used as the scientific description and the poster carries the visual summary.
Representative trimethoprim analogues are modelled in the cofactor-bound EcDHFR pocket for comparable trajectory analysis.
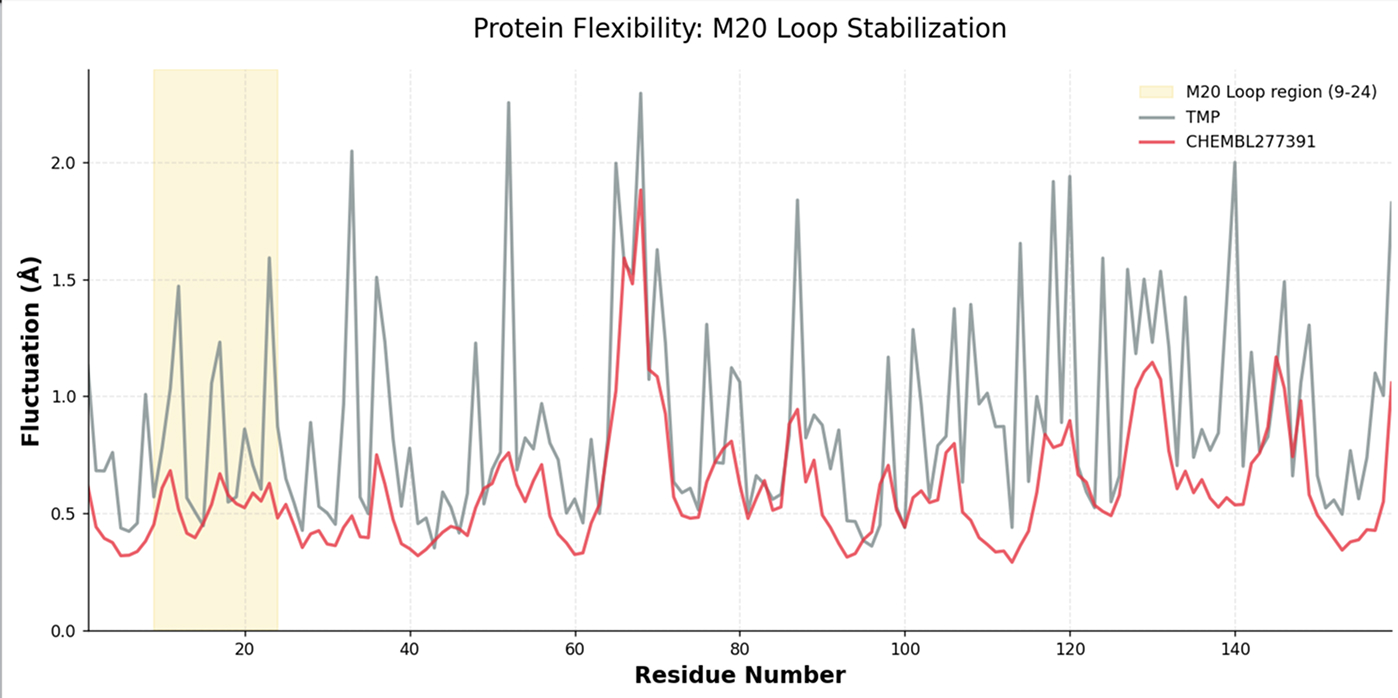
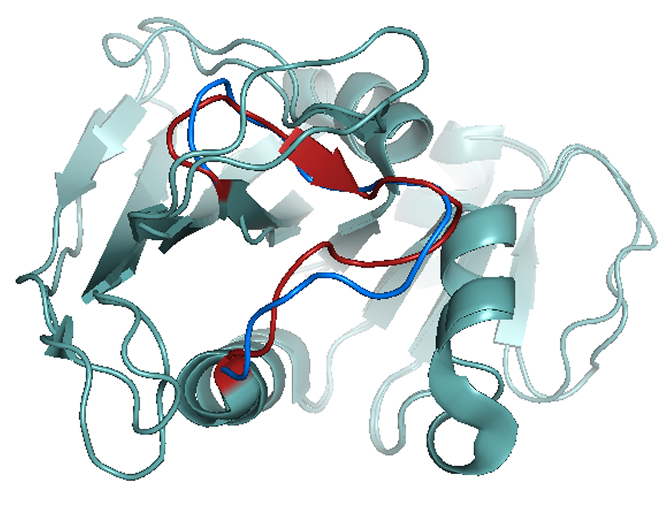
Trajectory descriptors capture binding-mode persistence, residue-level flexibility, and recurring pocket states.
Biased sampling probes conformational transitions that short unbiased simulations may under-sample.
Energetic and electronic descriptors expose effects omitted by docking scores and classical force fields.
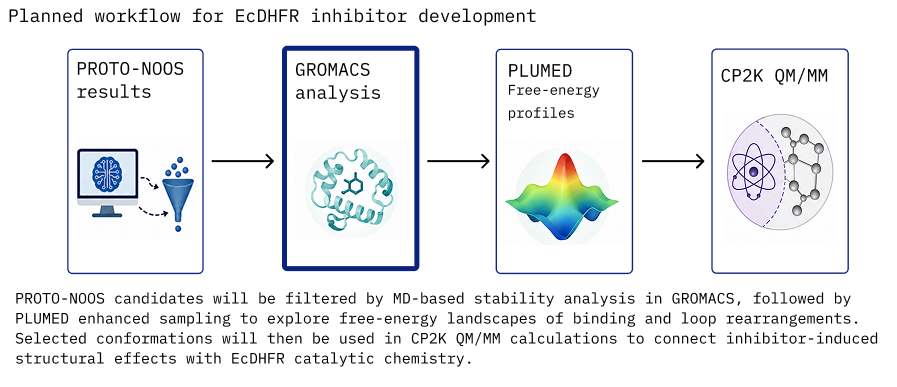
The available graphics summarise the biocomplex-analysis branch: system preparation, conformational sampling, energetic comparison, and electronic descriptors for data products that can feed later machine-learning refinement.
Connects structural modelling, MD, enhanced sampling, binding energetics, and electronic-structure analysis.
Tracks how ligand identity changes binding-pocket geometry, local flexibility, and conformational state occupancy.
Uses representative conformations to organize contact, energetic, and electronic descriptors into a reusable dataset.
CP2K-derived local electronic descriptors capture charge redistribution absent from classical docking scores.
The poster is the current citable visual artifact for this branch. A manuscript link can be added here once the text exists.